Using Rcompadre with the tidyverse
Patrick Barks & Owen Jones
2026-04-23
Source:vignettes/a02_RcompadreTidy.Rmd
a02_RcompadreTidy.RmdIntroduction
Rcompadre includes methods for a variety of functions in the “Tidyverse”, a popular group of R
packages geared toward data analysis, including dplyr (a grammar of data
manipulation) and ggplot2
(a grammar of graphics). This vignette covers manipulation of
CompadreDB objects using dplyr, and a few
examples of plotting CompadreDB objects using
ggplot2. Users new to these packages may wish to check out
more introductory materials (e.g. the dplyr vignette, or
the Data
Visualization chapter of R for Data Science).
Introduction to piping
We can take advantage of piping. The pipe (%>%) (from
the magrittr package),
passes an object on the left to the first argument of a function on the
right. There is also a native pipe (after R v. 4.1.0) but the
magrittr pipe has some advantages because it has a
placeholder element for when one does not want the left hand side result
to go to the first argument of the right hand expression.
Though we generally don’t need to, we can explicitly refer to the
object on the left using a dot (".").
The dot notation is particularly helpful if we want to pass the object on the left to an argument other than the first one.
x <- 1:3
y %>% data.frame(col1 = x, col2 = .) # use dot to pass object to second argumentUsing a pipe in a single line isn’t that helpful. The real benefits come when we use pipes in multi-line expressions to carry out a sequence of related operations.
Piping with Rcompadre
Let’s say we want to remove all rows where matA contains missing
values (NA). We can use cdb_flag() to add a
column checking for NA (column “check_NA_A”), and then use
filter() to remove those rows. Here are two approaches
without piping:
# approach 1 (nested functions)
compadre_use <- filter(cdb_flag(Compadre), check_NA_A == FALSE)
# approach 2 (intermediate step)
compadre_flag <- cdb_flag(Compadre)
compadre_use <- filter(compadre_flag, check_NA_A == FALSE)and here’s the equivalent piped sequence:
compadre_use <- Compadre %>%
cdb_flag() %>% # first argument is Compadre, from previous line
subset(check_NA_A == FALSE) # first argument is output of cdb_flag()The advantage of piping here is that we don’t have to use nested
functions (filter(cdb_flag())), and we don’t have to create
object names for every intermediate step in our analysis. An additional
alternative is to use subset() instead of
filter().
The mutate function
The mutate() function in dplyr adds one or more new
columns to a data frame (or in our case, a CompadreDB
object). Often the new column will be based on some transformation of
one or more of the existing columns.
Let’s say we want to conduct an analysis comparing Nordic countries
to the rest of Europe. We can use filter() to limit the
database to Europe, and then use mutate() to create a new
column identifying rows from Nordic countries.
compadre_euro <- Compadre %>%
filter(Continent == "Europe") %>%
mutate(Nordic = Country %in% c("NOR", "SWE", "DNK", "ISL", "FIN"))Using mutate with Rcompadre functions that return
vectors
A variety of Rcompadre functions take a
CompadreDB object as their first argument and return a
vector. To use these functions within mutate() in a piped
sequence, we generally need to explicitly refer to the object on the
left side of the pipe with a dot (".").
Here’s an example with the Rcompadre functions
mpm_has_active() and cdb_id_studies(), each of
which take a CompadreDB object and return a vector.
compadre_use <- Compadre %>%
mutate(has_active = mpm_has_active(.)) %>%
filter(has_active == TRUE) %>%
mutate(StudyID = cdb_id_studies(.))In the example above, the pipe passes a CompadreDB
object to the first arguments of mutate() and
subset(), respectively. The dot for the first argument is
implicit. Then, within mutate(), we use the dot again
(explicitly this time) to pass the CompadreDB object to
mpm_has_active() or cdb_id_studies().
We can also use the dot notation approach to extract components from
the CompadreMat objects in the column mat,
such as matA, matU, matF,
matC, matrixClass,
MatrixClassOrganized, or MatrixClassAuthor.
Each of these components can be accessed using an accessor function of
the same name. Here’s how to extract a list of matU and a
list of MatrixClassOrganized from each row of the database,
and add these lists to a CompadreDB object as new
columns:
compadre_unnest <- Compadre %>%
mutate(
mat_U = matU(.),
m_class_organized = MatrixClassOrganized(.)
)Vectorizing within mutate with the apply functions
Just like creating a new column with $
(e.g. compadre$new_col <- ...), each expression within
mutate() must return a vector of the same length as the
number of rows in the data frame, or return a single value which will be
recycled for all rows. For some operations this requires
vectorization.
Let’s say we want to calculate the population growth rate for every
matrix in the database. We can use the eigs() function in
the popdemo
package, but it can only take a single matrix at a time.
To apply eigs() to a list of matrices we can use
sapply(). But first we need to remove matrices with missing
values, because these will cause eigs() to fail.
compadre_lambda <- Compadre %>%
cdb_flag() %>%
filter(check_NA_A == FALSE) %>% # remove matrices with missing values
mutate(mat_A = matA(.)) %>% # extract list-column of matA
mutate(lam = sapply(mat_A, popdemo::eigs, what = "lambda"))
#> Warning: There was 1 warning in `dplyr::mutate()`.
#> ℹ In argument: `lam = sapply(mat_A, popdemo::eigs, what = "lambda")`.
#> Caused by warning in `FUN()`:
#> ! More than one eigenvalues have equal absolute magnitudeIn the example above, sapply() returns a scalar value
for every row of the database. If we instead want to derive a more
complex object for every row such as a vector or matrix, we can use the
function lapply() which always returns a list. In the
example below we use lapply() to calculate vectors of
stage-specific survival (column sums of matU) for every row
of the database.
Vectorizing over multiple arguments
Let’s say we want to know the survival probability for the first
‘active’ stage class (i.e. a stage that’s not dormant or propagule) for
every row of the database. One approach is to write our own function
that takes matU and the integer index of the first active
stage class, and returns the corresponding survival probability,
e.g.
SurvFirstActive <- function(matU, first_active) colSums(matU)[first_active]Notice that this function has two arguments, both of which will vary
across rows of the database. We therefore need to vectorize this
function over both arguments, which we can do with mapply()
(i.e. “multivariate apply”). Here’s an example:
compadre_surv_first_active <- Compadre %>%
mutate(
surv_1 = mapply(
FUN = SurvFirstActive, # function
matU = matU(.), # argument 1
first_active = mpm_first_active(.)
) # argument 2
)The group_by and summarize functions
The group_by() and summarize() functions
are used for split-apply-combine operations, where we want to apply a
function separately to different groups within our data, and then
recombine the results.
Here’s an example. We’ll use group_by() to group our
database by unique values of species (column
SpeciesAccepted), and then use summarize() to
count the number of unique populations (column
MatrixPopulation) for each group. Notice that the output is
a regular tibble (i.e. no longer a CompadreDB object) with
one row for each unique ‘group’ (i.e. species).
# count number of unique populations by species
Compadre %>%
group_by(SpeciesAccepted) %>%
summarize(n_populations = length(unique(MatrixPopulation))) %>%
arrange(desc(n_populations)) # arrange in descending order of n_pops
#> # A tibble: 110 × 2
#> SpeciesAccepted n_populations
#> <chr> <int>
#> 1 Aster amellus 3
#> 2 Echinacea angustifolia 3
#> 3 Lepidium davisii 3
#> 4 Pyrrocoma radiata 3
#> 5 Androsace elongata 2
#> 6 Armeria caespitosa 2
#> 7 Asarum canadense 2
#> 8 Astragalus peckii 2
#> 9 Astragalus scaphoides 2
#> 10 Calochortus lyallii 2
#> # ℹ 100 more rowsIf, instead of a summary table with one row per species, we would
rather append the number of populations per species as a new column of
our CompadreDB object, then we can follow
group_by() with mutate() instead of
summarize(). Here’s an example:
# subset to species with 10+ unique populations
compadre_replicated_pops <- Compadre %>%
group_by(SpeciesAccepted) %>%
mutate(n_pops = length(unique(MatrixPopulation))) %>%
ungroup() %>%
subset(n_pops >= 10)Make sure to use ungroup() after you’re finished with
the groups, to prevent unexpected behaviour later on.
Obtaining a single representative of each species
It is a common task in comparative analyses to obtain a single
representative of each species. One way that users can do that is by
using group_by(), followed by slice() and
sample() to randomly select a representative from each
accepted species.
Using CompadreDB objects with ggplot
Rcompadre includes methods that allow CompadreDB objects to be used
as data arguments within ggplot(), just like any old data
frame. Below are some example plots.
Plot study coordinates on world map:
If the optional maps package is not installed, the world
map example below will be shown but not executed during vignette
rendering.
ggplot2::ggplot(Compadre, aes(Lon, Lat)) +
borders(database = "world", fill = "grey80", col = NA) +
geom_point(col = "steelblue", size = 1.8, alpha = 0.8)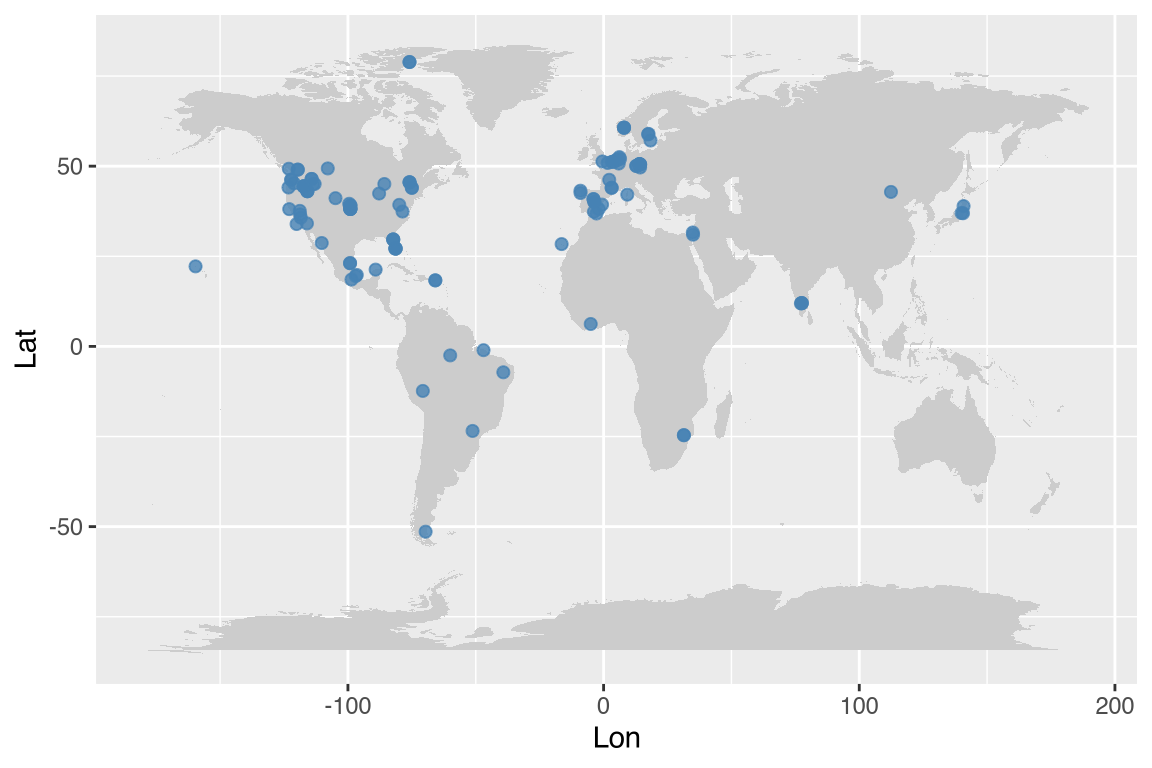
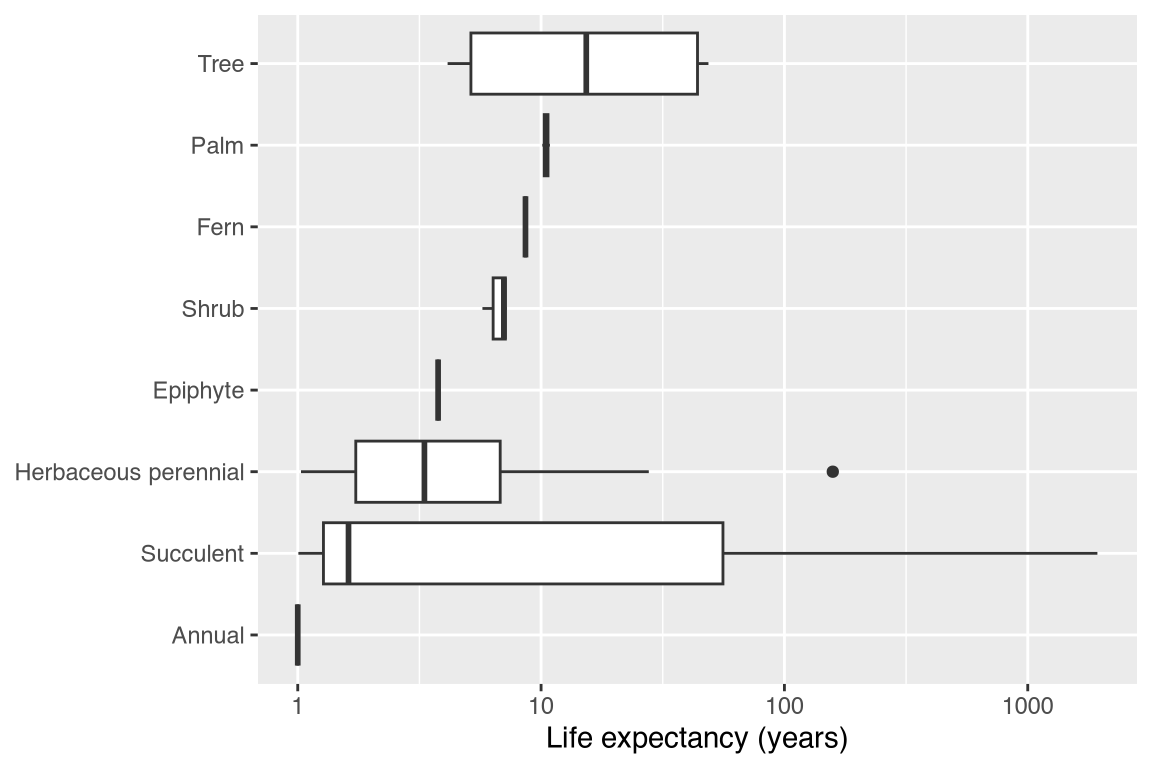
Boxplots of life expectancy from the first non-propagule stage, arranged by OrganismType, based on ‘grand mean matrices’ from studies of wild, unmanipulated populations:
This one is rather advanced, combining all of the topics in this vignette. So don’t worry if you can’t immediately understand every line.
# function to calculate life expectancy
lifeExpectancy <- function(matU, startLife) {
N <- solve(diag(nrow(matU)) - matU)
return(colSums(N)[startLife])
}
compadre_life_expect <- Compadre %>%
filter(
MatrixComposite != "Seasonal", # filter is the dplyr version of subset
MatrixTreatment == "Unmanipulated",
MatrixCaptivity == "W",
ProjectionInterval == "1"
) %>%
mutate(StageID = cdb_id_stages(.)) %>%
cdb_collapse(columns = "StageID") %>%
cdb_flag() %>%
filter(
check_NA_U == FALSE,
check_zero_U == FALSE,
check_singular_U == FALSE
) %>%
mutate(matU = matU(.), start_life = mpm_first_active(.)) %>%
mutate(life_expectancy = mapply(lifeExpectancy, matU, start_life)) %>%
filter(life_expectancy >= 1) %>%
mutate(OrganismType = reorder(OrganismType, life_expectancy, median))
ggplot2::ggplot(compadre_life_expect, aes(OrganismType, life_expectancy)) +
geom_boxplot() +
scale_y_log10() +
coord_flip() +
labs(x = NULL, y = "Life expectancy (years)")